Software für Ihre Technische Dokumentation
Welche Vorteile bietet die Software Technische Dokumentation?
Lassen Sie manuell geführte Dokumentationen zu Dossiers der Vergangenheit angehören – Verwalten Sie diese stattdessen vollkommen digital.
Lassen Sie unaufgeräumte Fileserver mit Mehrfachablagen und redundanten Ordnerstrukturen der Vergangenheit angehören.
Schützen Sie Ihre Daten vor unerwünschten Handlungen nicht autorisierter Benutzer sowie vor zerstörerischen Kräften und machen Sie sich keine Gedanken mehr über drohenden Datenverlust.
Lassen Sie die Module nach Ihren Wünschen und Vorlieben konfigurieren um die Einarbeitung Ihrer Kollegen zu erleichtern.
Erstellen und aktualisieren Sie digital Ihre Technische Dokumentation im Rahmen der Anforderungen der Medical Device Regulation.
Minimieren Sie die Prozesslaufzeiten für die Erstellung, Überarbeitung und Freigabe Ihrer Dossiers und reduzieren Sie überflüssigen Zeitaufwand zum Suchen und Zusammenstellen der aktuellen Dokumente.
Nutzen Sie d.velop documents (ehemals d.3ecm) als Ihre zentrale Informationsplattform auf der Sie Ihre gesamten Daten kurzfristig abrufen können.
Greifen Sie jederzeit auf Ihre Daten zu. Dabei spielt es keine Rolle an welchem Ort Sie sich befinden.
Entdecken Sie einige Features der Technischen Dokumentation
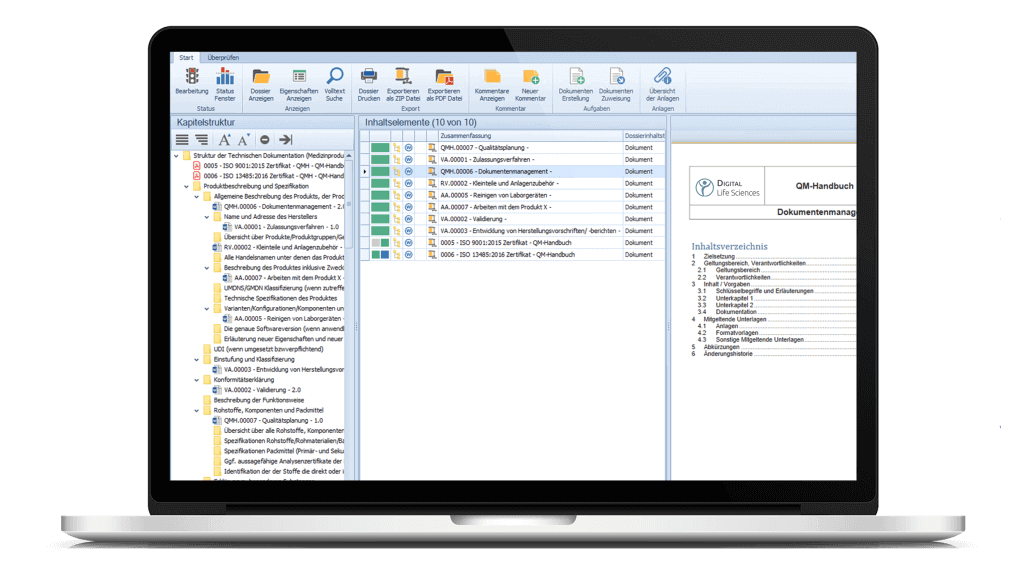
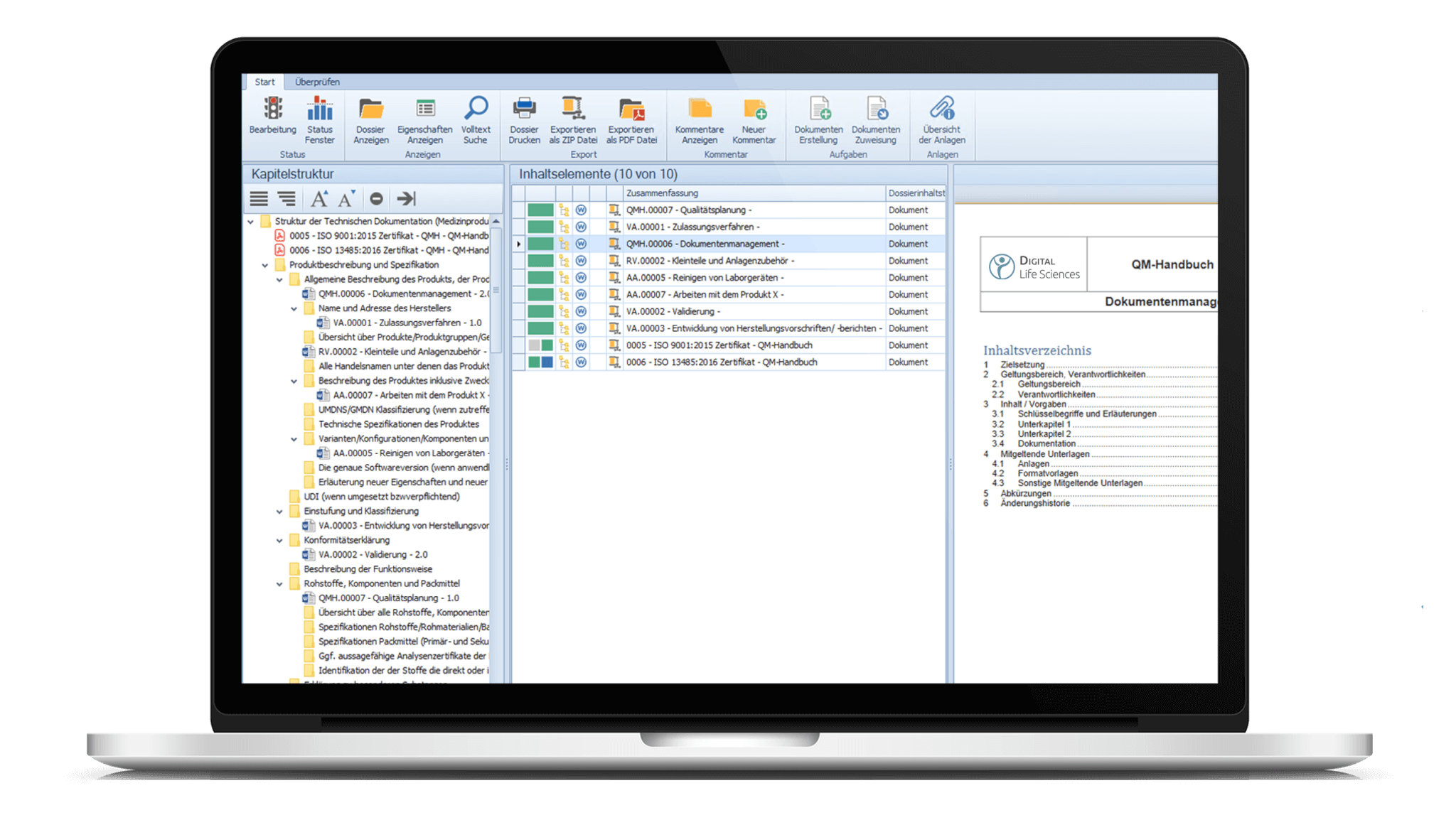
Mittels der Dossiervorlage können Dokumente aus dem eDMS/eQMS bereits in die Struktur übernommen werden. Fügen Sie weitere Dokumente und Anlagen der Dossieranwendung hinzu und ordnen Sie diese den jeweiligen Kapiteln per Drag & Drop zu.
Drucken Sie Ihre Dossiers oder exportieren Sie diese als ZIP-Datei oder PDF/A. Es besteht zudem die Möglichkeit lediglich Teile bzw. bestimmte Kapitel des Dossiers zu drucken und/oder zu exportieren.
Über die Drop Zone können gelenkte und nicht gelenkte Dokumente per Drag & Drop dem Dossier hinzugefügt werden. Dokumente können auch direkt den jeweiligen Knoten/Kapiteln im Dossier via Drag & Drop zugeordnet werden.
Zu jedem Dokument kann eine Vorschau innerhalb des flexibel anpassbaren Lesebereiches angezeigt werden.
Erstellen Sie Vorlagen und Kapitelstrukturen für Ihre Dossiers gemäß Ihren Bedürfnissen und legen Sie Verantwortlichkeiten für die einzelnen Bereiche fest. Dossier-Templates, z. B. für die MDR-Baumstruktur, sind automatisch enthaltener Auslieferungsgegenstand bei der Einführung des Moduls.
Wünschen Sie einen Live-Einblick zur Software?
Erhalten Sie in nur 45 Minuten einen Live-Einblick in die Möglichkeiten der Abbildung eines prozessorientierten Dossiermanagements anhand eines Musteranwendungsfalles. Erfahren Sie, wie unsere Lösung die Erstellung und Aktualisierung Ihrer Technischen Dokumentation im Rahmen der Anforderungen der Medical Device Regulation (MDR) unterstützen und vereinfachen kann.
Auszug einiger Features der Technischen Dokumentation
- Vorlagenmanagement für unterschiedliche Strukturen Ihrer Dossiers
- Aufgabenmanagement für die Bearbeiter einzelner Kapitel eines Dossiers (Workflow)
- Nahtlose Integration in unser Modul Dokumentenlenkung
- Vollständigkeitsprüfung Ihrer Dossiers
- Kontrolle des aktuellen Bearbeitungsstands Ihrer Dossiers
- Versionierung Ihrer Dossiers und einfacher Zugriff auf alle alten Versionsstände
- Priorisierbare Kommentare
- Export des gesamten Dossiers oder ausgewählter Abschnitte als PDF- oder ZIP-Datei
- Druck des Gesamtdossiers oder selektierter Bereiche des Dossiers
- Integrierte elektronische und GxP-konforme Signatur sowie Audit-Trail Funktion
- Klassifizierung der Dossiers über Attribute im ECM d.velop documents (ehemals d.3ecm)
Regulatorische Anforderungen der Technischen Dokumentation
- ISO 13485 MPG
- Medical Device Regulation (MDR) — EU-Richtlinie MDR 2017/745
33 Gute Gründe für eine Zusammenarbeit mit Digital LS
Sie sind noch nicht überzeugt? Erfahren Sie 33 Gute Gründe, die für eine Zusammenarbeit mit der Digital Life Sciences GmbH sprechen. Dabei zeigen wir Ihnen sowohl Gründe aus Anbietersicht, aus Softwaresicht sowie weitere allgemeine Gründe, die uns auszeichnen.
Kundenmeinung zur Technischen Dokumentation
“Wir arbeiten seit Jahren sehr erfolgreich und zufrieden mit der d.velop AG zusammen. Auf der Suche nach einem zuverlässigen Partner für die technische Dokumentation unserer gesamten Medizinprodukte sind wir auf den Life Sciences Client gestoßen. Mit der Umstellung auf die neue Struktur der EU MDR konnten wir das Potenzial vom Life Sciences Client für uns vollständig nutzen und somit die Arbeit bei der Dokumentenerstellung erleichtern und die Akzeptanz im Unternehmen verbessern.”

Das könnte Ihnen auch gefallen …
Die Technische Dokumentation ist ein Baustein der Digital Life Sciences Lösungssuite. Jedes Produkt für sich ist schon leistungsstark, gemeinsam eingesetzt sind sie sogar noch besser.

Dokumentenlenkung Software
Ob Arbeits- / Verfahrensanweisung (SOP’s), Prozessbeschreibungen, Prüfvorschriften oder andere Dokumenttypen — alle erstellen, revisionieren und unterschreiben Sie digital mit der Software Dokumentenlenkung.

Schulungsmanagement Software
Erweitern Sie das Modul “Dokumentenlenkung” um die aktive Planung und Protokollierung der Qualifikationen Ihrer Mitarbeiter mit unserer Schulungsmanagement Software.
QM-Prozesse (Complaint | DC | CAPA | CC)
Digitalisieren Sie Ihre ISO-Prozesse. Steuern Sie Ihre produktionsbegleitenden QM-Prozesse mittels digitaler Workflows.
Häufig gestellte Fragen (FAQ’s) zur Technischen Dokumentation
Gibt es Templates für die Struktur des Dossiers?
Können Kapitelstrukturen angepasst werden?
Wie stelle ich sicher, dass ein Mitarbeiter alle relevanten Dokumente in das Dossier einfügt?
Kann ich Dokumente aus dem File-System im Dossier verwenden?
Können mehrere Mitarbeiter an einem Dossier arbeiten?
Können Dokumente mehrfach verwendet werden?
Warum nutzt man die Technische Dokumentation zur Zulassung eines Medizinproduktes?
Werden die Dokumente im Dossier bei neuen Versionen aktualisiert?
Bei Freigabe eines Dossiers wird die Version ‚eingefroren‘, Änderungen sind nur in einer Folgeversion möglich. Mit der Übersicht ‚Versionsstände‘ erhalten Sie eine Übersicht der Dokumentenversionen. Um neue Versionen zu übernehmen, erstellen Sie eine neue Version des Dossiers und entscheiden, welche Version der Dokumente sie übernehmen.
Bei Dokumenten, die dem Dossier als Anlage hinzugefügt wurden, wird immer die gültige Freigabeversion verwendet.
Können einzelne Dokumente aus dem Dossier gedruckt werden?
Ist ein Teilexport des Dossiers möglich?
Sind die aus dem Dossier exportierten Daten geschützt?
Wird der Export protokolliert?
Welche Dateiformate können im Dossier verwendet werden?
Wie kann unsere Software Sie bei der technischen Dokumentation unterstützen?
Unsere Technische Dokumentation Software ist eine Anwendung, die primär für die Verwaltung von technischen Dokumentationen (“Dossiers”) verwendet wird. Sie kann dabei eine Vielzahl von Dossiers verwalten. Die Software erlaubt Ihnen zum Beispiel die Erstellung einer technischen Dokumentation gemäß MDR (EU Medizinprodukte-Verordnung). Das Ziel ist hierbei die revisionssichere Verwaltung sämtlicher Dokumente und Inhalte, die im Leben eines Medizinproduktes anfallen.
Wie Sie zukünftig als Anwender von unserer Software für die Technische Dokumentation in Ihrem Unternehmen profitieren können, zeigen wir Ihnen gerne in einem Live-Webcast zur Software.
Übersicht Medical Device Regulation (MDR)
Medical Device Regulation (MDR) — Verordnung 2017/745 (EU) über Medizinprodukte
Die Medizinprodukteverordnung (MDR) ist seit dem 26. Mai 2021 in der Europäischen Union (EU) gültig. Dieser Artikel bietet Informationen zu ihrem Inhalt und den Umsetzungsbestimmungen für die Medizintechnikbranche.
EU-Verordnung maßgeschneidert auf den Bereich der Medizintechnik
Die “Verordnung (EU) 2017/745 des Europäischen Parlaments und des Rates über Medizinprodukte” trat bereits am 5. April 2017 in Kraft. Sie tritt an die Stelle der bisherigen “Richtlinie 93/42/EWG über Medizinprodukte”. Konkret bedeutet dies für den Bereich der Medizintechnik, dass die EU-Verordnung 2017/745 folgende bisherige Richtlinien ablöst:
- Richtlinie 93/42/EWG über Medizinprodukte (MDD);
- Richtlinie 90/385/EWG, aktive implantierbare Medizinprodukte — Active Implantable Medical Devices (AIMDD).
Die EU-Gremien entschieden sich für eine separate Regelung in Bezug auf die Richtlinie 98/79/EG über In-vitro-Diagnostika (IVD). Diese Richtlinie wurde nicht in die Medizinprodukteverordnung übernommen. An ihre Stelle tritt die separat formulierte EU-Verordnung “In-vitro Diagnostic Medical Devices Regulation 2017/746 (IVDR)”.
Verbindlich in ganz Europa seit dem 26. Mai 2021
Im Gegensatz zur abgelösten Richtlinie hat die neue Verordnung des EU-Parlaments internationale Bedeutung erlangt. Ihre verbindliche Gültigkeit erstreckt sich ab dem Stichtag 26. Mai 2021 auf alle EU-Mitgliedstaaten. Es ist zu beachten, dass bis Oktober 2019 noch keine nationalen Rechtsakte der einzelnen Mitgliedsstaaten beschlossen wurden. Dies gilt auch für deutsche Besonderheiten, Anforderungen und zu erlassende Vorschriften. Es liegt zwar ein entsprechender Entwurf für ein deutsches Durchführungsgesetz vor, doch rechnet das Bundesgesundheitsministerium mit Übergangsfristen.
Gemäß dem vorliegenden Gesetzestext gilt bei der Umsetzung eine Übergangsfrist von drei Jahren (26.05.17 — 25.05.21*). Spätestens seit dem Ablauf der Frist sind Hersteller verpflichtet, ein MDR-Zertifikat vorzulegen. Nur so ist es ihnen erlaubt, ein Produkt erstmalig auf den Markt zu bringen.
*Im Zuge der COVID-19-Pandemie wurde der Geltungsbeginn der Verordnung auf den 26. Mai 2021 verschoben.
Ziel und Notwendigkeit der neuen EU-Verordnung
Im Kern zielt die neue Verordnung auf eine optimierte, einheitliche Regelung für die Markteinführung von Produkten der Medizintechnik ab. Im Fokus stehen dabei die Faktoren Produktsicherheit und Produktqualität. Für Marktteilnehmer und Anwender besteht die neue EU-Verordnung aus einer Mischung aus bekannten Inhalten der früheren Richtlinie 93/42/EWG und weitreichenden Neuerungen. Zu den wesentlichen inhaltlichen und verfahrenstechnischen Aspekten für Sie und Ihre Branche gehören die folgenden Inhalte der Brüsseler Verordnung:
Regeln zur Klassifizierung von Medizinprodukten
Neue Begriffe und Regeln führen zu Veränderungen bei der Zuordnung bestehender Produkte zu teilweise geänderten Produktklassen. Von diesen Veränderungen ist beispielsweise der Produktbereich der “Stand-alone”-Software betroffen.
Verfahren zur Konformitätsbewertung
In Anlehnung an das Regelwerk für die Klassifizierung unterscheidet sich nun auch das Konformitätsbewertungsverfahren je nach Klasse.
Technische Dokumentation/Dokumenten Management System
Die technische Dokumentation nach der Medizinprodukteverordnung erweist sich als wesentlich detaillierter. Vorteile bei der Kontrolle und Dokumentation für Unternehmen werden deutlich, wenn man sich das neu zugrundeliegende, tabellarisch strukturierte Schema im Detail ansieht. Die Unterteilung in einzelne Kategorien in direkter Verknüpfung mit den zugehörigen detaillierten Anforderungen bildet die Grundlage für eine detaillierte technische Dokumentation.
Klinische Bewertungen/klinische Prüfungen
Deutlich erhöhte Anforderungen kennzeichnen diesen Aktionspunkt. Dies gilt insbesondere für risikoreiche Produktgruppen.
Firmenverantwortung/Sicherheitsbeauftragter
Eine wesentliche Änderung für Ihr Unternehmen ergibt sich aus der Forderung der EU-Gremien nach einer verantwortlichen Person, die für die Einhaltung der regulatorischen Anforderungen in ihrer Organisation zuständig ist.
Qualitätsmanagement
Die FDA Medical Device 21 CFR 820 (FDA = U.S. Food and Drug Administration) formuliert die Anforderungen an Managementsysteme von Medizinprodukteherstellern. In dieser Funktion ist Device 21 CFR 820 ein Pendant zur ISO 13485. Kernanforderung ist, dass Verfahrensanweisungen wie Dokumentenlenkung, Beschaffung, Entwicklung sowie Produktion dokumentiert und analog umgesetzt werden.
Marktüberwachungsmechanismen
Insbesondere die verpflichtende Nutzung der EUDAMED-Datenbank sorgt für mehr Transparenz. Sie steht sowohl der Öffentlichkeit als auch den direkten Wettbewerbern als Informationsmedium zur Verfügung. Durch entsprechende Verknüpfungen werden die Merkmale jedes Medizinprodukts abgebildet. Auch die Rückverfolgung von Lieferketten ist möglich
Wege zur Einhaltung der Medizinprodukteverordnung
Im Interesse der rechtzeitigen Einhaltung der neuen Verordnung sind Sie die verantwortliche Person im Unternehmen. Ein zeitlich und inhaltlich stimmiger Plan für die Umstellung Ihres Unternehmens auf die Medizinprodukteverordnung ist Voraussetzung für Ihren Erfolg. Sie sollten den Lebenszyklen Ihres Medizinprodukteportfolios hohe Priorität einräumen. Bestehende MDD-Zertifizierungen verlieren nach den EU-Vorschriften am 26. Mai 2024 ihre Gültigkeit. Bitte bedenken Sie bei Ihrer Planung, dass Ihre bereits nach MDD zertifizierten Produkte auch das neu zu schaffende Konformitätsverfahren nach MDR durchlaufen müssen. Ein “Grandfathering” ist nicht garantiert.
In der Phase der Umstellung sind Sie in Ihrer Funktion als Hersteller oder Lieferant besonders gefordert. Oberste Priorität hat die Benennung und Schulung der Person, die in Ihrem Unternehmen für die Einhaltung der Vorschriften verantwortlich ist (MDR Artikel 15). Aktualisierungen der internen Qualitätssicherungsverfahren, der Dokumentation und der geänderten Produktklassifizierung sind zwingend erforderlich.
Vor diesem Hintergrund wird das folgende standardisierte Vorgehen nach den folgenden 4 Punkten empfohlen:
- Durchführung der Konformitätsbewertung in Bezug auf das Produkt
- Ausstellung der Konformitätserklärung
- “CE”-Kennzeichnung der Produkte
- Registrierung des Unternehmens und der Produkte in der EUDAMED-Datenbank.
Fazit und Kritik zur neuen Verordnung
Mit der MDR haben sich die EU-Behörden vorgenommen, eine moderne, einheitliche Lösung für das Qualitätsmanagement und die Marktfreigabe von Medizinprodukten zu schaffen. Der Qualität und Sicherheit der Produkte wird dabei große Aufmerksamkeit geschenkt. Vielleicht gehören auch Sie zu den kleinen und mittleren Unternehmen der Medizintechnikbranche, die eine Überregulierung durch das umfangreiche Regelwerk und zusätzliche Kontrollstellen befürchten?
Insbesondere die Bewertung der Konformität durch den einzustellenden “Compliance Manager” dürfte in der Startphase zu einem personellen Problem werden. Im Härtefall könnte ein Mangel an zertifizierten Prüfern die für die Marktfreigabe notwendige Zertifizierung verzögern. Medizintechnische Industrieverbände äußern die Befürchtung, dass sich ein Engpass bei den Zertifizierungen negativ auf die Markteinführung innovativer und marktfähiger Produkte auswirken könnte.
Schnellkontakt
Sie haben eine Frage zur Technischen Dokumentation?
Unser Vertriebsteam hilft Ihnen zeitnah und gerne weiter.
